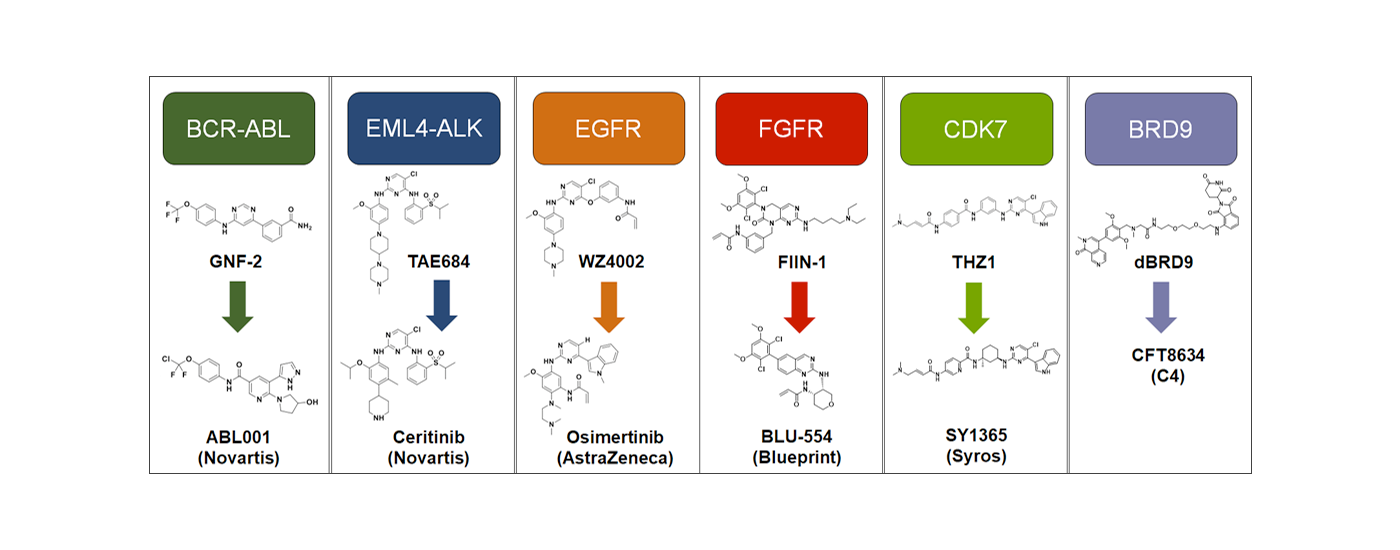
Inhibitors of protein kinases are now a well-established class of drugs, especially in oncology. Despite high response rates in kinase-addicted tumors, resistance to monotherapy inevitably arises. We are currently developing innovative approaches that target kinases in a way that makes resistance less likely to occur. Our arsenal includes covalent inhibitors, allosteric inhibitors, and small molecule degraders.
Some example projects include:
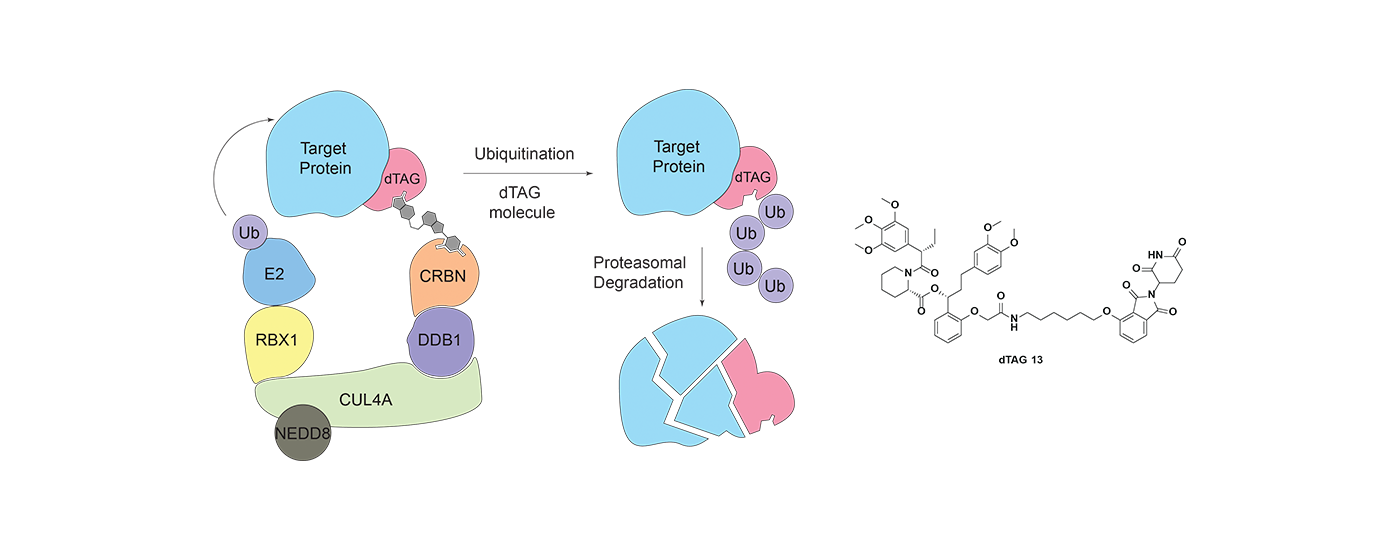
Heterobifunctional small molecule degraders are an innovative chemical approach, whereby rather than inhibiting protein function, proteins are selectively targeted for degradation by the proteasome. We are synthesizing small molecule degraders that hijack various E3 ligases such as cereblon (CRBN) and VHL, and developing systematic approaches to evaluate effects of degrader molecules. We are also developing cutting-edge technology platforms that enable targeted degradation of any protein of interest.
Our recent accomplishments include:
![]()
Gene expression networks function to maintain stable cell states; changes in gene expression output are instructive for organism development and disease. Simple physical closeness, or proximity, between DNA-sequence-specific transcription factors (TFs) and epigenetic regulators inside a cell can be sufficient for gene regulation. Using our understanding of epigenetic regulation and molecular biology-inspired pharmacological assays, we developed molecules that induce proximity between endogenous TFs and epigenetic regulators, called Transcriptional Chemical Inducers of Proximity (TCIPs). These compounds rewire gene expression and produce new cell fates without introducing genetic modifications to the cell or organism.
Initial efforts focused on rewiring the repressive TF BCL6, which is deregulated in lymphoma. BCL6 epigenetically silences pro-apoptotic genes, permitting lymphoma cells to proliferate. Our recent accomplishments include:
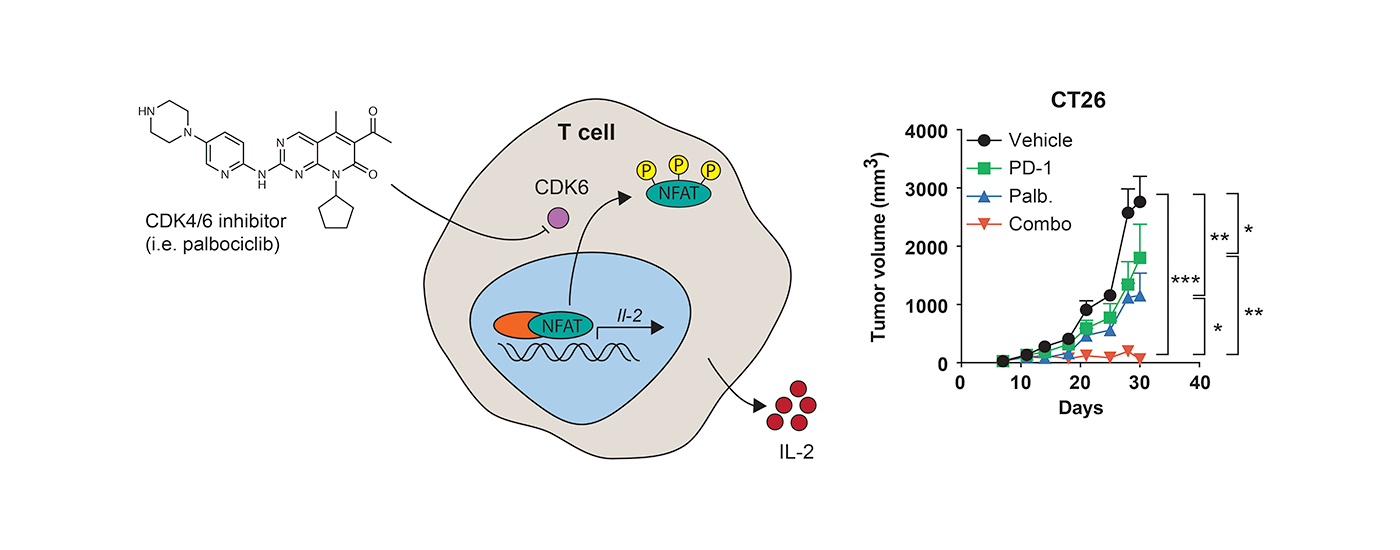
Immunotherapy is becoming a mainstay of cancer treatment. The most effective agents currently in use are antibodies that block receptors involved in immune evasion including PD-1, PDL1 and CTLA4. We have demonstrated that small molecule mediated targeting of intracellular proteins, such as CDK4/6, enhances the ability of the immune system to recognize and destroy tumor cells. We continue our efforts to discover additional intracellular targets in the context of immunooncology.
Our key insight in this area is:
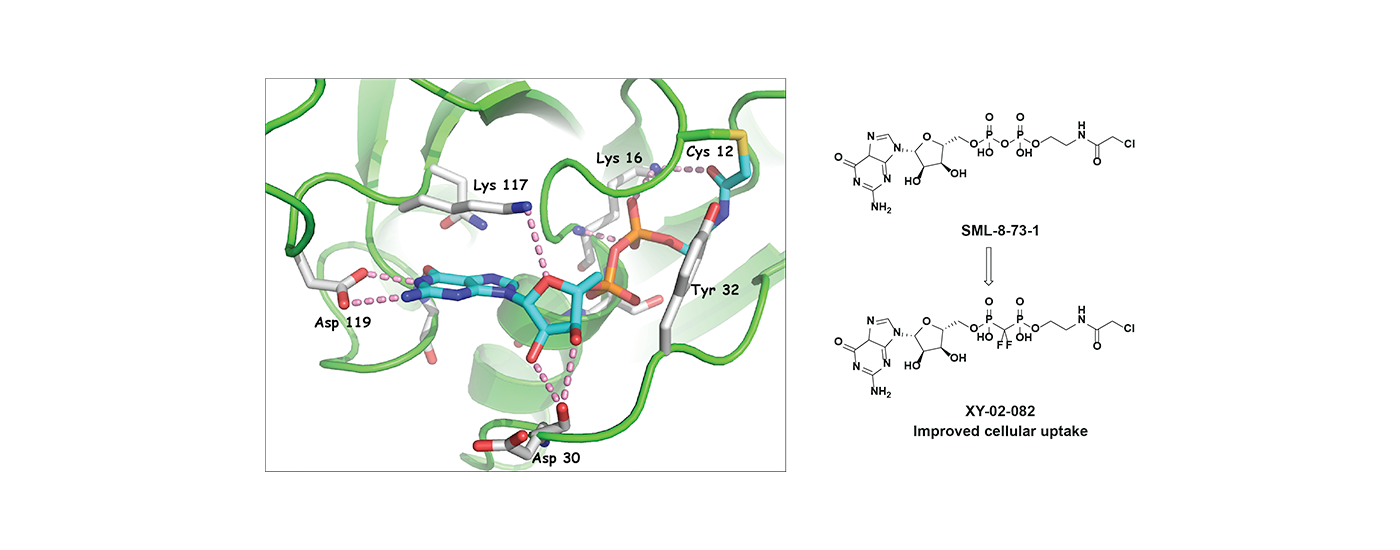
Challenging Drug Targets: Mutant K-RAS is a frequent genetic alteration found in many cancers that is yet to be effectively drugged. We are exploring novel approaches to targeting mutant K-RAS using a guanosine diphosphate mimetic covalent inhibitor. We are also exploring the development of small molecule degraders of HER3, a catalytically inactive member of the EGFR-family that has been successfully drugged with antibodies but for which no small molecule antagonists are currently known.
Some examples of our work in this area are:

Despite intensive research efforts, much of the kinome is poorly annotated and incompletely understood. This ‘dark’ kinome represents a significant portion of the kinase space and an untapped targeting opportunity. We are developing a suite of approaches to tackle these understudied kinases including the development of selective inhibitors, tagging strategies for rapid degradation, and phosphoproteomics approaches to enable identification of kinase substrates.
Our efforts in this area include:
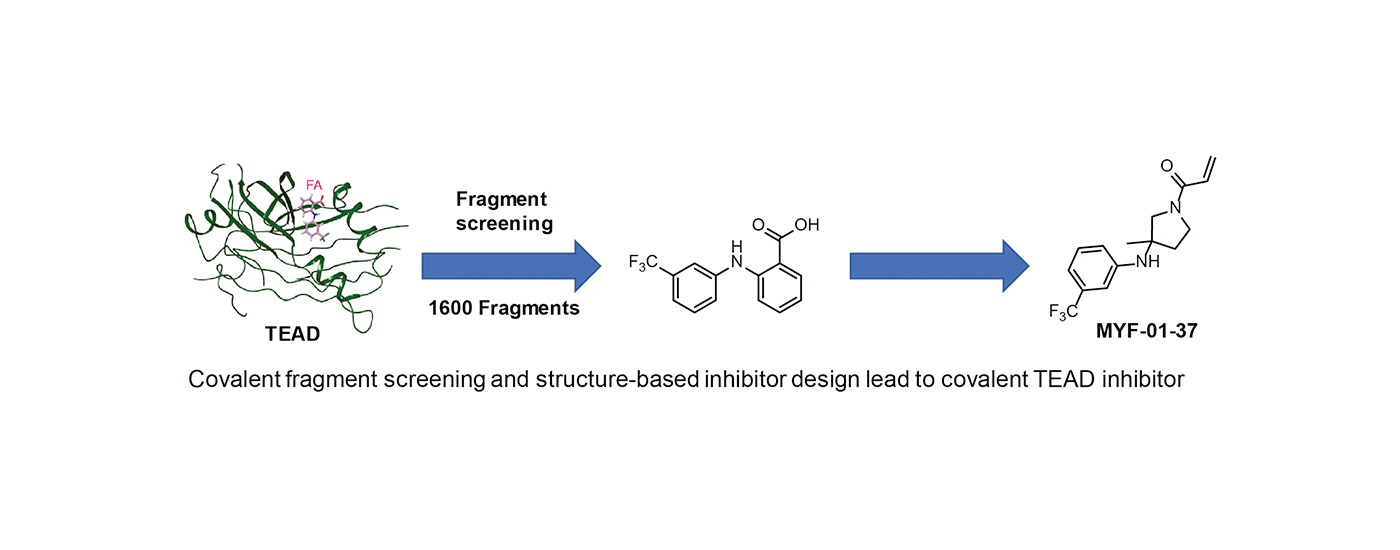
Covalent inhibitors have traditionally come from natural products (ie betalactams, aspirin) or from structure-based inhibitor design (ie covalent kinase inhibitors such as osimertinib). Recent advances in quantitative mass spectrometry are enabling proteome-wide surveys of thousands of proteins bearing reactive cysteines. We are developing procedures that incorporate covalent fragment screening coupled with structure-based inhibitor design to develop first-in-class covalent probes for previously difficult-to-drug targets. Two recently published examples are covalent PIN1 inhibitors (sulphopin) and covalent TEAD inhibitors.
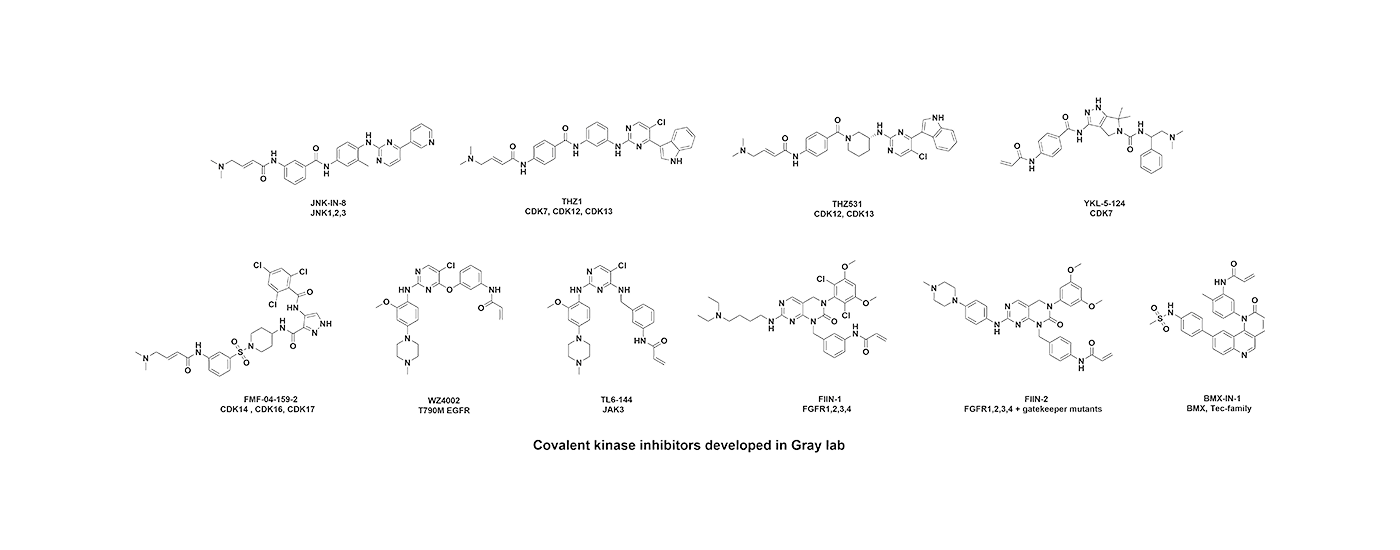
Developing selective ATP-competitive kinase inhibitors can be challenging due to the highly conserved nature of the ATP binding site in the approximately 580 human kinases. We have worked to develop approaches for making selective kinase inhibitors that form a covalent bond with cysteine residues that are frequently found in and around the ATP-binding pocket. Covalent kinase inhibitors have the advantage of being able to combine selectivity obtained from non-covalent recognition of specific scaffold with careful positioning of a reactive group adjacent to a particular cysteine residue. Moreover, covalent inhibitors can achieve full target engagement in vivo often without the need for an extensive campaign to modulate the pharmacokinetic properties of the compound. For kinase inhibitors that are obligately covalent to achieve potency, mutation of the cysteine can provide a chemical-genetic method for proving that the observed pharmacology is ‘on-target’. Over the years, we have developed numerous selective covalent kinase inhibitors and have developed chemical proteomic methods for assessing their cellular selectivity.
